El análisis de microarray cromosómico (CMA), también conocido como array de hibridación genómica comparativa o array de CGH, es una prueba diagnóstica que permite detectar desequilibrios cromosómicos clínicamente significativos (aneuploidías) y variaciones submicroscópicas del número de copias (microdeleción/microduplicación) en todo el genoma.
El análisis de microarray cromosómico es el gran referente para la detección de deleciones y duplicaciones en todo el genoma.
El CMA ofrece resolución submicroscópica para visualizar pequeñas regiones que el cariotipado no es capaz de detectar. La sensibilidad del CMA se basa en el número de sondas moleculares utilizadas y en su cobertura del genoma. Cuanto mayor sea el número de sondas (180K, 400K, 750K…), mayor será su sensibilidad.
El CMA puede detectar:
Pequeñas microdeleciones y duplicaciones cromosómicas
Variaciones en el número de copias (VNC)
Anomalías cromosómicas numéricas (aneuploidías)
Reordenamientos desequilibrados
Exceso de homocigosidad; dependiente de la plataforma
Riesgo estimado de heredar enfermedades recesivas o trastornos de impronta; dependiente de la plataforma
Triploidía y tetraploidía; dependiente de la plataforma
El CMA puede ofrecerse en la fase de preconcepción si se dan una o varias de las siguientes indicaciones:
Si un niño afectado previamente se presenta con un cariotipo o WES normales, pero se sospecha un trastorno genético.
Para comprobar si una variación en el número de copias que se ha detectado en el niño afectado es de novo o heredada de sus padres.
Siempre se recomienda realizar un cariotipo constitucional de ambos progenitores si se sospecha un reordenamiento cromosómico equilibrado (p. ej. abortos recurrentes, infertilidad, etc.)
El CMA se ofrece en la infancia o la edad adulta si se dan una o varias de las siguientes indicaciones; el microarray cromosómico puede valorarse como prueba de primera línea en estos casos:
Si una persona o un feto presentan múltiples anomalías congénitas que no son específicas de un síndrome genético bien identificado
Si un cariotipo es negativo y el fenotipo de la persona indica la existencia de aneuploidía cromosómica
Si una persona o un feto presentan un retraso del desarrollo aparentemente no sindrómico o discapacidades intelectuales
Si una persona presenta trastornos del espectro autista
Si un feto tiene una malformación o se produce un mortinato de etiología desconocida
El CMA puede utilizarse para ofrecer un diagnóstico en casos donde otras pruebas han fallado si se dan específicamente uno o varios de los siguientes síntomas o trastornos:
El CMA puede ofrecerse en un producto de concepción si ha habido un caso de abortos espontáneos
Informes y resultados de CMA
Hay varios tipos diferentes de CMA disponibles en Igenomix:
CMA HD (la prueba más sensible)
Sondas del número de copias (1,9 millones) + SNP (750K).
Cobertura de todo el genoma.
Puede detectar regiones de baja heterocigosidad, disomía uniparental (DUP), bajo nivel de mosaicismo y heterogeneidad de las muestras.
La mayor densidad de sondas.
CMA 750K (la prueba más económica)
Sondas del número de copias solo (550K) + SNP (200K).
Énfasis en las regiones clínicamente relevantes.
Identificación de regiones de excesiva homocigosidad indicadoras de DUP, puede sugerir consanguinidad y determinar genes candidatos para pruebas ulteriores.
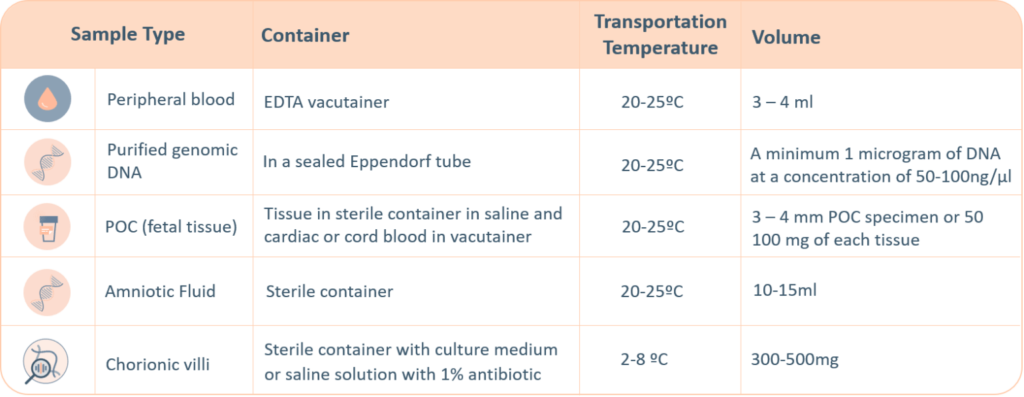
Requisitos de la muestra del CMA
Las pruebas genéticas de Igenomix aceptan los siguientes tipos de muestras. Se sugiere realizar un etiquetado meticuloso del tubo con información de identificación única; un etiquetado incorrecto puede causar el rechazo de la muestra. La información mínima requerida para identificar y aceptar una muestra es: nombre completo del paciente, fecha de nacimiento, sexo y número de expediente médico.
La muestra de sangre materna debe enviarse con todas las muestras de productos de concepción, VC y amnióticas.
Se dará preferencia a todas las muestras prenatales..
El formulario de consentimiento informado y el formulario de solicitud del test (incluidos en el kit suministrado) deben cumplimentarse debidamente, llevar la firma del paciente y enviarse con las muestras en la caja de envío o por correo electrónico al laboratorio. Igenomix le enviará todos los documentos necesarios para la recogida y el transporte del kit correspondiente a nuestro laboratorio.
Metodología del CMA
Las deleciones inferiores a 50 kb y las duplicaciones inferiores a 400 kb no se pueden revisar. Se indican las variaciones en el número de copias (VNC) detectadas si tienen relevancia clínica evidente o presunta; no se indicarán las VNC sin contenido genético relevante o presentes comúnmente en la población general. Las regiones de homocigosidad se indican cuando un tramo largo contiguo de homocigosidad (LCSH) es mayor de 8-15 Mb (dependiendo de la ubicación cromosómica y de la probabilidad de un trastorno de impronta), o cuando la proporción total de LCSH autosómico es superior al 3 % (para esta estimación solo se considera LCSH autosómico mayor de 3 Mb). Las posiciones lineales genómicas se indican en relación a la versión 37 de NCBI (hg19).
Los resultados de la prueba se interpretan de acuerdo con las recomendaciones y directrices de ISCA (por sus siglas en inglés, Consorcio de Normas Internacionales para Arreglos Citogenómicos) descritas a continuación:
Un resultado positivo indica que se ha identificado una variación del número de copias en conexión con el fenotipo de enfermedad bajo estudio. Este escenario permitirá ofrecer asesoramiento genético u orientación personal sobre posibles tratamientos médicos, la progresión de la enfermedad, las estrategias reproductivas o de prevención y las implicaciones potenciales para otros miembros de la familia.
Un resultado negativo indica que no se ha identificado una variación en el número de copias causante de enfermedad en la prueba realizada. Esto no garantiza que la persona esté sana o no tenga otros trastornos genéticos o médicos. Además, un resultado negativo no descarta una causa genética de la enfermedad ni elimina el riesgo para la futura descendencia. No obstante, si se obtiene un resultado negativo en la prueba y se sabe que la variante en cuestión está presente en miembros familiares afectados, esto descarta un diagnóstico de ese trastorno genético en el sujeto de prueba. Un resultado negativo puede explicarse por varias causas, incluidos un conocimiento genético limitado y limitaciones asociadas a la metodología utilizada.
El hallazgo de una variante de significado incierto indica que se ha detectado una variación en el número de copias (VNC), pero que actualmente se desconoce si esa VNC está asociada con un trastorno o enfermedad genética. Una variante de significado incierto no es lo mismo que un resultado positivo y no aclara si el sujeto de prueba tiene un riesgo mayor de desarrollar un trastorno o enfermedad genética.
El cambio podría ser una variante genética normal o podría causar una enfermedad. En este caso, puede ser recomendable realizar análisis ulteriores, incluida una prueba a ambos progenitores y a otros miembros familiares afectados o no afectados. En ocasiones, es necesario realizar pruebas auxiliares para identificar el fenotipo que presenta el sujeto de prueba. También puede requerirse el historial médico o información detallada de otros miembros familiares para ayudar a aclarar el resultado.
La interpretación de los resultados se basa en la información disponible actualmente en la literatura médica, la investigación y las bases de datos científicas. Puesto que la literatura médica y el conocimiento científico cambian constantemente, la nueva información disponible en el futuro puede reemplazar o complementar la información utilizada por Igenomix para interpretar los resultados. Normalmente no se realiza un nuevo análisis de los resultados de informes anteriores para tener en cuenta nuevas evidencias, pero es una opción posible bajo solicitud.
Este sitio web almacena cookies en su ordenador. Estas cookies se utilizan para recopilar información acerca de la forma en que usted interactúa con nuestro sitio web y recordarlo. Usamos esta información para personalizar y mejorar su experiencia de navegación y para realizar análisis y recuento de los visitantes, tanto en este sitio web como a través de otros medios.